Schwachstelle für
Herzproblem
entdeckt
Forscher des Deutschen Zentrums für Herzinsuffizienz Würzburg (DZHI) machen fehlenden Calciumkanal als Auslöser für Arrhythmien und Herzinsuffizienz beim seltenen Barth-Syndrom aus.
Edoardo Bertero (links) und Michael Kohlhaas an der Single-Cell-Force-Anlage im DZHI.
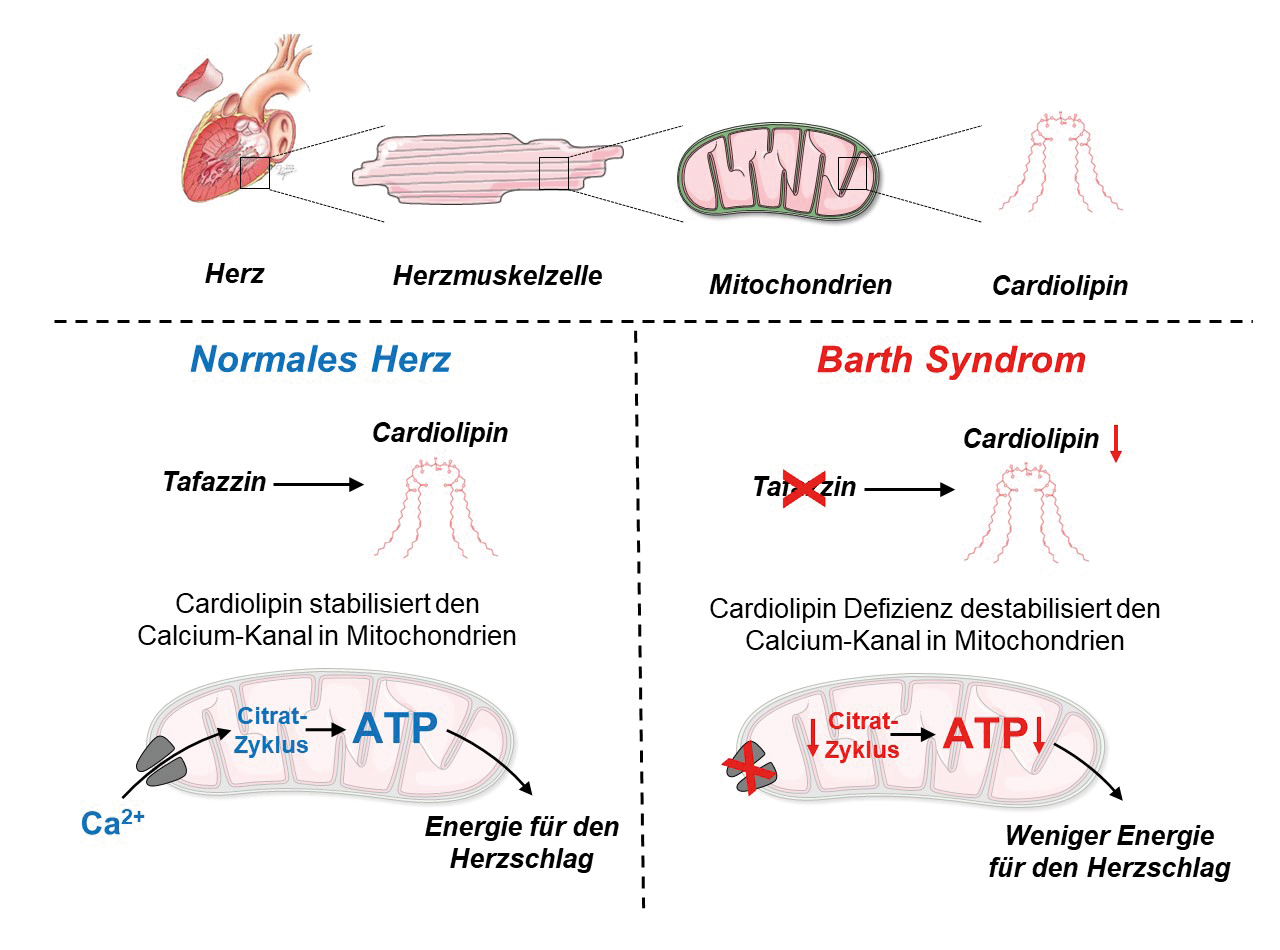
Unser Herz pumpt pro Minute vier bis fünf Liter Blut in unseren Körper, bei hoher Belastung sogar bis zu 30 Liter pro Minute, sofern es gesund ist. Bei Patientinnen und Patienten, die am Barth-Syndrom leiden, schlägt das Herz bei Anstrengung zwar schneller, die Pumpleistung, also der Auswurf, kann aber nicht entsprechend gesteigert werden. Die Folge dieser verminderten Herzfunktions-Reserve bei Belastung ist Luftnot. Hinzu kommen Herzrhythmusstörungen, die auch zum plötzlichen Tod führen können. Doch die Betroffenen dürfen möglicherweise bald aufatmen. Im DZHI hat Professor Dr. Christoph Maack mit seinem Team den Calciumkanal in den Mitochondrien als Ursache für ihre Herzfunktionsstörungen entlarvt.
Das Barth-Syndrom geht auf einen Defekt des Tafazzin-Gens zurück, und Tafazzin produziert Cardiolipin, einen wesentlichen Bestandteil der Mitochondrienmembran. Die Wissenschaftler fanden heraus, dass die durch den Defekt des Tafazzin-Gens beeinträchtige Energiegewinnung der Herzmuskelzellen mit dem Calciumhaushalt zusammenhängen. Durch die verminderte Calciumaufnahme in den Mitochondrien, den Kraftwerken der Herzmuskelzelle, wird die Aktivierung des Citratzyklus gestört. Im Citratzyklus werden mithilfe des energieliefernden Coenzyms NADH Elektronen für die Produktion des energiereichen Moleküls Adenosintriphosphat (ATP), und über NADPH Elektronen für die Entgiftung von Sauerstoffradikalen hergestellt. Durch fehlenden Calciumkanal leeren sich die Speicher Das DZHI-Department Translationale Forschung hat nun den Mechanismus erkannt, warum sich das Herz-Zeit-Volumen nicht steigern lässt, und warum vermehrt Arrhythmien auftreten. „Wir haben beobachtet, dass der Kanal, der für den Calciumimport in die Mitochondrien verantwortlich ist, der so genannte mitochondriale Calcium- Uniporter, kurz MCU, in Mäusen mit Tafazzin-Knockdown fast vollständig verschwunden war“, erklärt Edoardo Bertero. Dies sei wichtig für Patienten mit Barth-Syndrom, weil es erklärt, warum ihre Herzen nicht in der Lage sind, ihre Auswurfleistung bei körperlicher Anstrengung zu erhöhen; aber auch für die allgemeine Herzphysiologie, weil es eine bisher nicht gewürdigte Funktion von Cardiolipin aufdeckt, nämlich die Stabilisierung des MCU-Protein-Komplexes. Diese Entdeckung, die im renommierten Journal Circulation der American Heart Asscociaton publiziert wurde, könnte nicht nur ein wichtiger Therapieansatz bei der Behandlung des Barth-Syndroms sein, sondern auch bei anderen Herzerkrankungen mit erhaltener Pumpfunktion, und im speziellen bei anderen genetischen Kardiomyopathien. Autorin: Kirstin Linkamp Publikation im AHA Journal Circulation: Loss of Mitochondrial Ca2+ Uniporter Limits Inotropic Reserve and Provides Trigger and Substrate for Arrhythmias in Barth Syndrome Cardiomyopathy, Bertero et al. Originally published 14 Oct 2021 https://doi.org/10.1161/CIRCULATIONAHA.121.053755