
Pathomechanismen von spinalen Muskelatrophien
Unsere Arbeitsgruppe hat Zellkultursysteme zur Untersuchung dieser Pathomechanismen entwickelt, um Perspektiven für neue Therapien aufzuzeigen.
Spinale Muskelatrophie (SMA)
Die klassische Form der SMA beruht auf dem Verlust bzw. der Dysfunktion des sogenannten Survival-Motoneuron-Proteins (SMN). Für SMA gibt es zugelassene genetische Therapien, die auf die Hochregulation des SMN-Proteins abzielen. Dennoch ist das Wissen über den Krankheitsmechanismus, der zum Verlust der Motoneurone führt, noch lückenhaft (Jablonka et al., Neurol Res Pract 2022).
Untersuchungen an Tiermodellen haben ergeben, dass bei einer SMN-Defizienz die Degeneration der Motoneurone an deren Synapse der motorischen Endplatte (NMJ) beginnt. Dies ist verbunden mit einer verminderten Neurotransmitterausschüttung aufgrund einer defekten Kalzium- (Ca2+) Homöostase. Bei einer IGHMBP2-Defizienz beginnt die Motoneuronendegeneration hingegen primär im Zellkörper im Rückenmark, sodass die daraus folgende Muskelatrophie eher nicht durch eine defekte Neurotransmitterausschüttung verursacht wird.
Um solche in-vivo-Beobachtungen und Messungen besser zu interpretieren, nutzen wir primäre Motoneurone aus den entsprechenden Mausmodellen bzw. re-differenzierte Motoneurone aus induzierten pluripotenten Stammzellen (iPSCs).
Ca²⁺-Homöostase entscheidend für die Funktion und das Überleben von Motoneuronen
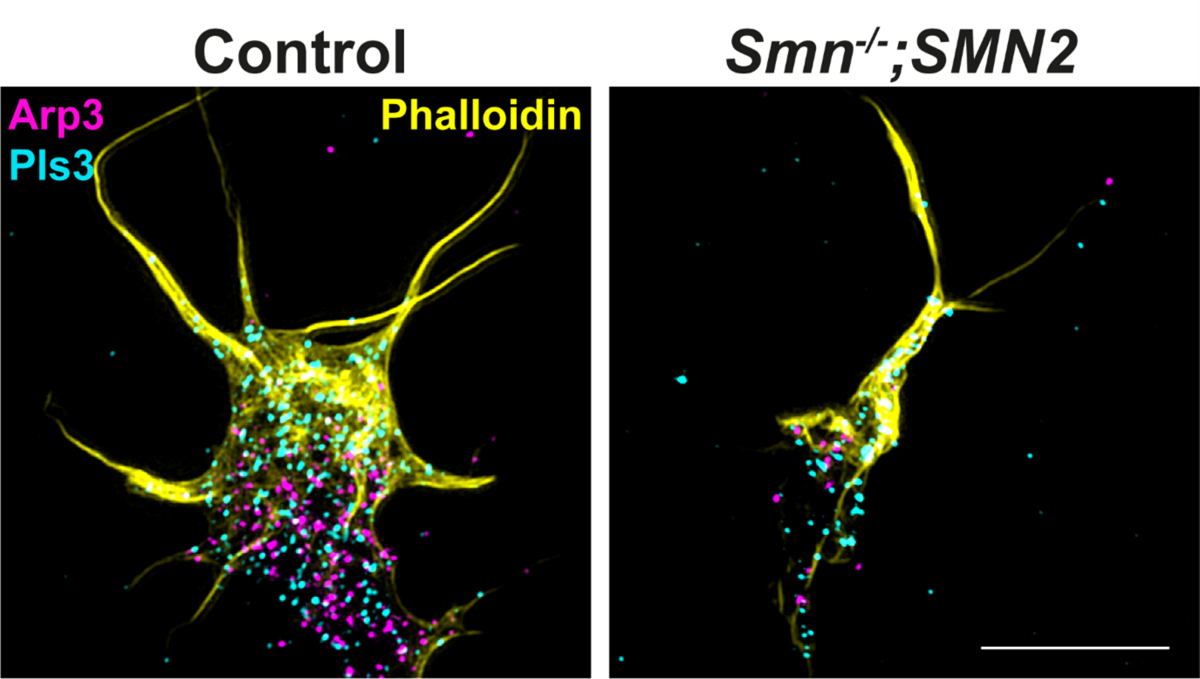
Mithilfe unserer Zellkultursysteme haben wir massive Veränderungen in der Ca2+ Homöostase festgestellt, die mit einem defekten Transport von mRNAs aus dem Zellkörper in das axonale Kompartiment von SMA-Motoneuronen verbunden sind (Hennlein et al., JCB 2023). Insbesondere sind Transkripte betroffen, die für Zytoskelettproteine wie Aktin-Isoformen, Komponenten des präsynaptischen Apparats für die Vesikelfreisetzung sowie den sogenannten SMA-Modifier Plastin 3 (PLS3) kodieren (Hennlein et al., JCB 2023). PLS3 hat eine modifizierende Wirkung auf die Bündelung von F-Aktin (siehe Abbildung 1). Dies hat wiederum Auswirkungen auf die korrekte Anordnung von Transmembranproteinen, wie den spannungsabhängige Kalziumkanälen (Cav2.1 oder Cav2.2) oder Rezeptoren für neurotrophe Faktoren (z.B. trkB/BDNF) (Hennlein et al., JCB 2023). Eine fehlerhafte Anordnung bzw. Lokalisation dieser Kanäle oder Rezeptoren beeinträchtigt die zelluläre Differenzierung von Motoneuronen und somit auch die Ausschüttung von Neurotransmittern, die für die Übertragung von Bewegungsimpulsen auf die Skelettmuskeln verantwortlich sind. Die verzögerte Motoneuronendegeneration, und das signifikant verlängerte Überleben von SMA-Mäusen durch den Cav2.1-Blocker/Modifier (R-Roscovitine) zeigen die zentrale Bedeutung einer balancierten Ca2+-Homöostase für die Aufrechterhaltung der Neurotransmission (Tejero et al., iScience 2020).
Aktuell untersuchen wir die Untereinheit Cacna2d2 des aktiven Zonen-assoziierten Kalziumkanals Cav2.1, der wiederum für eine effiziente Neurotransmission an der Motorendplatte unentbehrlich ist. Unser Ziel ist es zu zeigen, dass die genetische Modifikation von Cacna2d2 die Effizienz der Neurotransmission steigert und dem Krankheitsphänotyp entgegenwirkt.
Ausgewählte Publikationen
Hennlein L, Ghanawi H, Gerstner F, Palominos García E, Yildirim E, Saal-Bauernschubert L, Moradi M, Deng C, Klein T, Appenzeller S, Sauer M, Briese M, Simon C, Sendtner M, Jablonka S (2023). Plastin 3 rescues cell surface translocation and activation of TrkB in spinal muscular atrophy.
J Cell Biol. 222(3):e202204113.
Jablonka S, Hennlein L, Sendtner M (2022). Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders.
Neurol Res Pract. 2022 4(1):2.
Tejero R, Balk S, Franco-Espin J, Ojeda J, Hennlein L, Drexl H, Dombert B, Clausen J-D, Torres-Benito L, Saal-Bauernschubert L, Blum R, Briese M, Appenzeller S, Tabares L, Jablonka S (2020). R-Roscovitine improves motoneuron function in mouse models for Spinal Muscular Atrophy.
iScience 23:100826.
Spinale Muskelatrophie mit Ateminsuffizienz Typ 1 (SMARD1)
SMARD1 (Spinal Muscular Atrophy with Respiratory Distress Type 1) wird durch Punktmutationen im IGHMBP2-Gen verursacht, welches für die Ribosomen-assoziierte RNA-Helikase IGHMBP2 kodiert. Inzwischen wurden auch klinische Studien mit adeno-assoziierten Viren (AAV9) zur Hochregulation von IGHMBP2 bei SMARD1 begonnen (Jablonka et al., Biomedicines 2024).
Im Gegensatz zur SMN-Defizienz hat eine IGHMBP2-Defizienz hingegen keinerlei Auswirkungen auf die Ca2+-Homöostase von Motoneuronen (Surrey et al., Neuroscience 2018). Anhand von Tiermodellen konnte gezeigt werden, dass die Degeneration der Motoneurone nicht an der motorischen Endplatte beginnt – wie bei SMA. Vielmehr ist ein prä-symptomatischer Verlust der Motoneuronzellkörper im Rückenmark zu beobachten.
Beim SMARD1-Mausmodell der Nmd2J-Maus findet man neben der Muskelatrophie auch eine reine Myopathie des Diaphragmas und des Wadenmuskels. Dies kann durch die externe Applikation von IGF1 kompensiert werden (Krieger et al., Brain 2014). Transkriptom- und Proteom-Analysen von Ighmbp2-defizienten Motoneuronen konnten zeigen, dass die Expression von Extrazellulären-Matrix (ECM)- Proteinen in Ighmbp2-defizienten Motoneuronen signifikant betroffen ist. Gegenwärtig untersuchen wir die zellulären und molekularbiologischen Auswirkungen der ECM-Protein-Defizienz auf die Entwicklung der Motoneurone.
Ausgewählte Publikationen
Jablonka S, Yildirim E. (2024). Disease Mechanisms and Therapeutic Approaches in SMARD1-Insights from Animal Models and Cell Models.
Biomedicines 2024 Apr 11;12(4):845
Surrey V, Zöller C, Moradi M, Balk S, Lork AA, Dombert B, Saal-Bauernschubert L, Briese M, Appenzeller S, Fischer U, Jablonka S (2018). Impaired local translation of β-actin mRNA in Ighmbp2 deficient motoneurons: implications for spinal muscular atrophy with respiratory distress (SMARD1). Neuroscience 386:24-4
Krieger F, Elflein N, Saenger S, Wirthgen E, Rak K, Frantz S, Hoeflich A, Toyka K.V, Metzger F, Jablonka S (2014). PEGylated IGF-I delays motor function defects in a SMARD1 mouse model.
BRAIN 137:1374-1393.
