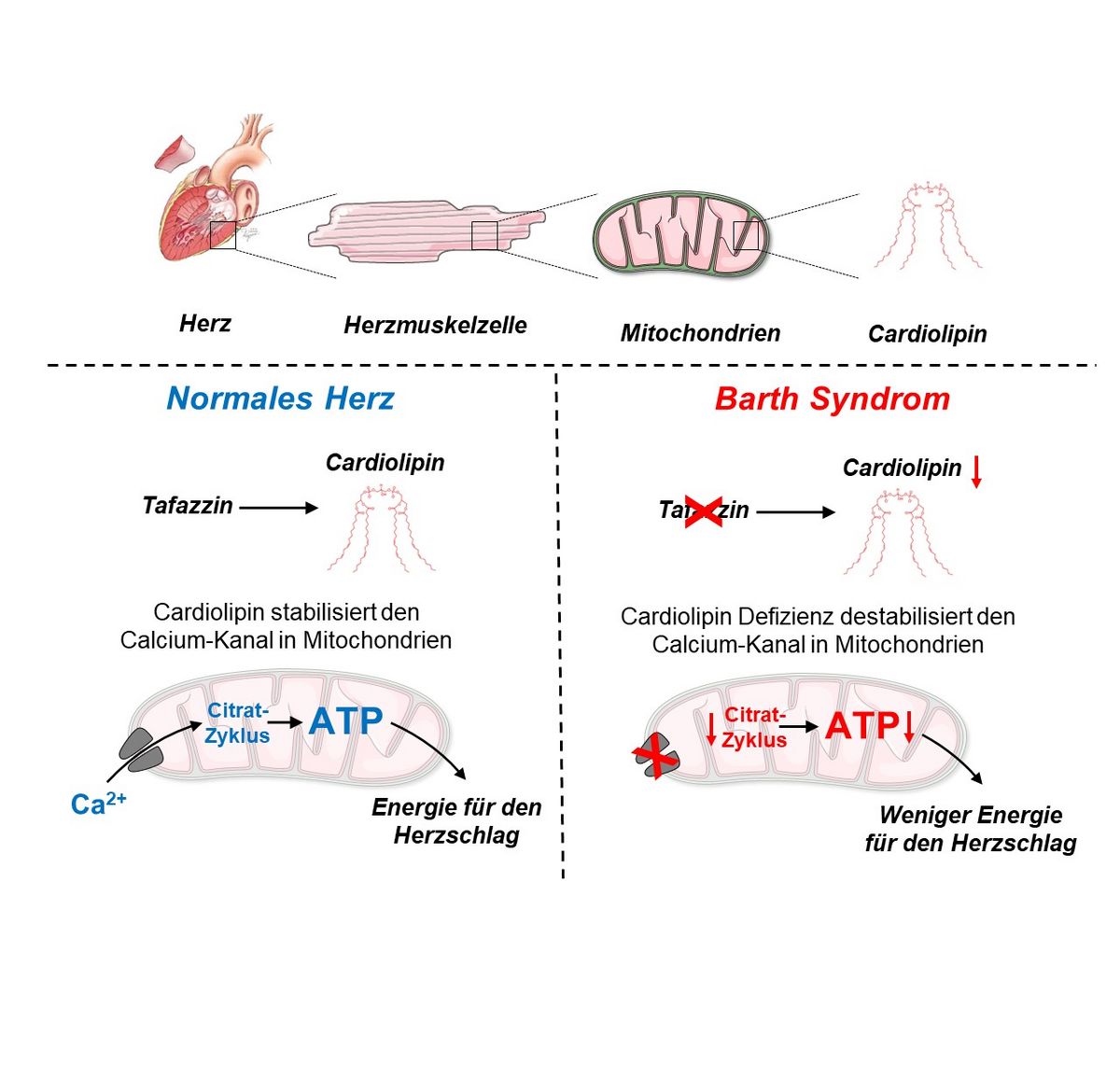
Patienten mit dem Barth-Syndrom dürfen möglicherweise bald aufatmen. Im Deutschen Zentrum für Herzinsuffizienz Würzburg (DZHI) hat Christoph Maack mit seinem Team den Calciumkanal in den Mitochondrien als Ursache für ihre Herzfunktionsstörungen entlarvt. Das Barth-Syndrom geht auf einen Defekt des Tafazzin-Gens zurück, und Tafazzin produziert Cardiolipin, einen wesentlichen Bestandteil der Mitochondrienmembran. Die Erkrankung betrifft meist Jungen im frühen Kindesalter und verursacht Herzschwäche und Herzrhythmusstörungen. Die Wissenschaftler fanden heraus, dass durch den Defekt des Cardiolipin der Calciumkanal in Mitochondrien verloren geht. Da Calcium der wichtigste Botenstoff für die Anpassung der Energieproduktion an einen erhöhten Bedarf ist, erklärt dieser Defekt die Unfähigkeit der Barth-Herzen, bei körperlicher Aktivität die Pumpleistung zu steigern, aber auch das Auftreten von Herzrhythmusstörungen. Diese Erkenntnisse, die jetzt im renommierten Journal Circulation der American Heart Association veröffentlicht wurden, sind nicht nur ein Lichtblick in der Behandlung des seltenen Barth-Syndroms, sondern könnten auch zum verbesserten Verständnis und der Behandlung der weiter verbreiteten Herzinsuffizienz mit erhaltener Pumpfunktion (HFpEF) beitragen.
Das Herz pumpt in der Regel pro Minute vier bis fünf Liter Blut in unseren Körper, bei hoher Belastung sogar bis zu 30 Liter pro Minute, sofern es gesund ist. Bei Jungen, die am Barth-Syndrom leiden, schlägt das Herz bei Anstrengung zwar schneller, der Auswurf kann aber nicht entsprechend gesteigert werden. Die Folge dieser verminderten Herzfunktions-Reserve bei Belastung ist Luftnot. Hinzu kommen Herzrhythmusstörungen, die auch zum plötzlichen Tod führen können.
Weniger Calcium = weniger Energie in Herzmuskelzellen
Der Kardiologe Christoph Maack und der Biologe Jan Dudek forschen bereits seit vielen Jahren an den Krankheitsmechanismen des Barth-Syndroms. Sie fanden heraus, dass die durch den Defekt des Tafazzin-Gens beeinträchtige Energiegewinnung der Herzmuskelzellen mit dem Calciumhaushalt zusammenhängen. Durch die verminderte Calciumaufnahme in den Mitochondrien, den Kraftwerken der Herzmuskelzelle, wird die Aktivierung des Citratzyklus gestört. Im Citratzyklus werden mithilfe des energieliefernden Coenzym NADH Elektronen für die Produktion des energiereichen Moleküls Adenosintriphosphat (ATP), und über NADPH Elektronen für die Entgiftung von Sauerstoffradikalen hergestellt.
Durch fehlenden Calciumkanal leeren sich die Speicher
Die Forscher aus dem DZHI-Department Translationale Forschung, allen voran Edoardo Bertero, Alexander Nickel und Michael Kohlhaas, haben nun den Mechanismus erkannt, warum sich das Herz-Zeit-Volumen nicht steigern lässt, und warum vermehrt Arrhythmien auftreten.
Früher ging man davon aus, dass das Fehlen von Cardiolipin vor allem der Atmungskette Probleme bereitet und Sauerstoffradikale die Zellen schädigen. Das Cardiolipin ist auch bei vielen anderen Herzkrankheiten durch oxidativen Stress geschädigt. Ein Mangel an diesem Phospholipid stört die Atmungskette, wodurch weniger Energie produziert wird. "Obwohl wir in unseren Studien auch eine moderate Störung der Atmungskette feststellen konnten, haben wir keine übermäßigen Mengen an Radikalen gemessen", erklärt Edoardo Bertero, der Erstautor der Studie. "Stattdessen haben wir beobachtet, dass der Kanal, der für den Calciumimport in die Mitochondrien verantwortlich ist, der so genannte mitochondriale Calcium-Uniporter, kurz MCU, in Mäusen mit Tafazzin-Knockdown fast vollständig verschwunden war. Dies ist wichtig für Patienten mit Barth-Syndrom, weil es erklärt, warum ihre Herzen nicht in der Lage sind, ihre Auswurfleistung bei körperlicher Anstrengung zu erhöhen; aber auch für die allgemeine Herzphysiologie, weil es eine bisher nicht gewürdigte Funktion von Cardiolipin aufdeckt, nämlich die Stabilisierung des MCU-Protein-Komplexes.“
Entdeckung führt zu besserem Verständnis des Barth-Syndroms
Maack fügt hinzu: „Die Gen- und Proteinstruktur des mitochondrialen Calciumkanals ist erst seit zehn Jahren bekannt. Das Barth-Syndrom ist die erste uns bekannte Erkrankung, bei der ein relevanter Defekt des MCU in Herzzellen deren Funktion nachhaltig beeinträchtigt.“ Mit dieser Entdeckung liefern die Forscher des DZHI einen wichtigen Therapieansatz, möglicherweise nicht nur bei der Behandlung des Barth-Syndroms, sondern auch bei anderen Herzerkrankungen mit erhaltener Pumpfunktion, und im speziellen bei anderen genetischen Kardiomyopathien. „Hilfreich könnte vielleicht die Gabe von SGLT2-Hemmern sein. Sie reduzieren das Natrium in der Zelle, dadurch wird weniger Calcium aus den Mitochondrien herausgeholt, die Energiespeicher bleiben länger voll, sodass das Herz bei erhöhter Belastung besser mithalten kann“, spekuliert Maack. Dies müsse aber erst noch untersucht werden. Weniger empfehlenswert seien Wirkstoffe, die die Pumpkraft des Herzens steigern, indem sie das Natrium erhöhen, wie zum Beispiel das seit Jahrzehnten verwandte Digitalis.
In der Vergangenheit wurden Jungen mit Barth-Syndrom oft nicht älter als drei Jahre. Sie starben an Herzversagen oder Infektionen. Aber mit einer verbesserten Diagnose und einer angemessenen medizinischen Behandlung und Überwachung aller Symptome ist die Überlebensrate und die Zukunft dieser Menschen viel besser. „Genau das motiviert mich und spornt mich an. Die Krankheit ist zwar selten. Registriert sind etwa 300 Fälle weltweit. Wir gehen jedoch von einer hohen Dunkelziffer aus. Und was zählt ist das Schicksal jedes einzelnen“, betont Maack.
Die Arbeiten entstanden in enger Zusammenarbeit mit zahlreichen anderen Gruppen in Homburg/Saar, Göttingen und Würzburg. Gefördert wurden die Forschungsarbeiten durch die Margret Elisabeth Strauß-Projektförderung der Deutschen Herzstiftung, die Deutsche Forschungsgemeinschaft (DFG), das Bundesministerium für Bildung und Forschung (BMBF), den European Research Council (ERC) sowie die US-amerikanische Barth Syndrome Foundation.
Weitere Informationen zur Erkrankung: www.barthsyndrome.org
Publikation in AHA Journal Circulation: Loss of Mitochondrial Ca2+ Uniporter Limits Inotropic Reserve and Provides Trigger and Substrate for Arrhythmias in Barth Syndrome Cardiomyopathy
Edoardo Bertero, Alexander Nickel, Michael Kohlhaas, Mathias Hohl, Vasco Sequeira, Carolin Brune, Julia Schwemmlein, Marco Abeßer, Kai Schuh, Ilona Kutschka. Christopher Carlein, Kai Münker, Sarah Atighetchi, Andreas Müller, Andrey Kazakov, Reinhard Kappl, Karina von der Malsburg, Martin van der Laan, Anna-Florentine Schiuma, Michael Böhm, Ulrich Laufs, Markus Hoth, Peter Rehling, Michaela Kuhn ,Jan Dudek, Alexander von der Malsburg, Leticia Prates Roma and Christoph Maack
Originally published14 Oct 2021 https://doi.org/10.1161/CIRCULATIONAHA.121.053755